Relativistic Thermodynamics at
IHP
(Institut Henri Poincaré, founded in 1928) : Louis de Broglie (1892-1987; Nobel 1929), Olivier
Costa de Beauregard (1911-2007)
Abdelmalek Guessous,
Jean Fronteau,
etc.
For surely the atoms did not hold council, assigning order to each,
flexing their keen minds with questions of places, motions and who goes where.
But shuffled and jumbled in many ways. In the course of endless time,
they are buffeted, driven along, chancing upon all motions and combinations.
At last they fall into such an arrangement as would create this Universe. Lucretius (99-55 BC)
De rerum natura
(2003-11-11) The Concept of Temperature
Zeroth law: Stochastic transfer of energy.
Consider the situation pictured above:
A vibrating piston is retained by a spring at one end of a narrow
cavity containing a single bouncing particle.
We'll assume that the particle and the piston only have
horizontal motions.
Everything is frictionless and undamped,
as would be the case at the molecular level...
If the center of mass of two bodies has a (vectorial) velocity
v, an exchange of a momentum
dp between them
translates into the following transfer of energy
dE, in an elastic collision
(this expression remains valid
relativistically).
dE = v . dp
We would like to characterize an ultimate "equilibrium", in which
the average energy exchange is zero whenever the piston
and the particle interact.
Let's call V the velocity of the piston, M its mass and x the elongation of the spring that
holds it. The force holding back the piston is
-Mw2 x for some
constant w, so that, in the absence of shocks with the
particle, the motion is a sinusoidal function of the time t and the total energy E
given below remains constant:
x = A sin (wt + f)
V = Aw
cos (wt + f)
E = ½ M ( V 2 +
w 2 x 2 )
When the particle, of mass m and velocity v, collides with the piston, we have:
V > v and
X + v t =
A sin (wt + f)
The first condition expresses the fact that the particle moves
towards the piston just before the shock.
The second relation involves a
random initial position X, uniformly distributed in the cavity.
Only the value of X modulo 2A
[i.e., the remainder when 2A is divided into X] is relevant,
and if we consider that the length of the cavity is many times greater than 2A,
we may assume that X is uniformly distributed in some interval of length 2A.
For an ideal gas, the thermodynamical temperature T can be defined
by the following formula, which remains true in the
relativistic case (provided
that the averaging denoted by angle brackets is understood to be at
constant time
in the observer's coordinates). This appears as equation 14-4 in the 1967
doctoral dissertation of Abdelmalek Guessous
(supervised by Louis de Broglie).
3 k T = <
( v - <v> ) .
( p - <p> ) >
In this, v and p are, respectively, the speed and momentum of each
individual (monoatomic) molecule.
(2003-11-09) The Second Law: Entropy Increases
Heat travels only from hot to cold. Carnot's principle.
In 1824,
Sadi Carnot
gave a fundamental limitation
of steam engines by analyzing the ideal engine now named after him,
which turns out to be the most efficient of all possible heat engines.
This result is probably best expressed with the fundamental
thermodynamical concepts which were fully developped
after
Carnot's pioneering work, namely internal energy (U)
and entropy (S).
Entropy (S) is an extensive quantity because the entropy of the whole
is the sum of the entropies of the parts.
However, entropy is not conserved as time goes on:
It increases in any non-reversible transformation.
Temperature as a function of Entropy
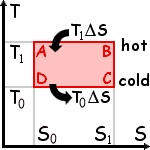
Carnot's Ideal Engine: The Carnot Cycle.
Hot (A to B) slow isothermal expansion.
The hot gas performs work and receives
a quantity of heat T1 DS.
Cooling (B to C) adiabatic expansion.
The gas keeps working without any exchange of heat.
Cold (C to D) slow isothermal compression.
The gas receives work and gives off (wasted) heat
T0 DS.
Heating (D to A) adiabatic compression from outside work (flywheel)
returns the gas to its initial state A.
As the internal energy (U) depends only on the state of the system,
its total change is zero in any true cycle.
So, the total work done by the engine in Carnot's cycle is equal to
the net quantity of heat it receives:
(T1-T0) DS.
For any simple hydrostatic system, that quantity would be the area enclosed
by the loop which describes the system's evolution in the above S-T diagram
(which gives the system's supposedly uniform temperature
as a function of its entropy).
This same mechanical work is also the area of the corresponding loop in the V-p
diagram (pressure as a function of volume).
This latter viewpoint may look more practical,
but it's far more obscure when it comes to discussing efficiency limits...
Efficiency of a Heat Engine
Carnot was primarily concerned with steam engines
and the mechanical power which could be obtained from the fire heating up
the hot reservoir (the cold reservoir being provided from the surroundings
at "no cost", from a water stream or from atmospheric air).
He thus defined the efficiency of an engine as the ratio of the work done
to the quantity of heat transferred from the hot source.
For Carnot's ideal engine, the above shows that this ratio boils down to
the following quantity, known as Carnot's limit :
1 -
T0 / T1
The unavoidable "waste" is the ratio of the extreme temperatures involved.
Refrigeration Efficiency
The primary purpose of a refrigerator (or an air-conditioning unit)
is to extract heat from the cold source
(to make it cooler). Its efficiency is thus usefully defined as the
ratio of that heat to the mechanical power used to produce the transfer.
So defined, the efficiency of a Carnot engine driven (backwards) as a refrigerator is:
( T1 / T0
- 1 ) -1
This is [much] more than 100%, except for
extreme refrigeration, which would
divide by two or more the ambient temperature above absolute zero.
The rated efficiency of commercial
cooling units (the "coefficient of performance"
COP) is somewhat lower, because it's defined in terms of the electrical
power which drives the motor (taking into account any wasted electrical energy).
Efficiency of a Heat Pump
A heat pump is driven like a refrigeration unit,
but its useful output is the heat transferred to the hot side
(to make it warmer).
A little heat comes from the electrical power not converted into
mechanical work, the rest is "pumped" at an efficiency which always exceeds
(by far) 100% of the mechanical work. For a Carnot engine, this latter
efficiency is:
(2006-09-12) What's worth knowing about a thermodynamical system
The two flavors of state variables: extensive and intensive.
Thermodynamics is based on the statement (or belief) that almost all details
about large physical systems are irrelevant or impossible to describe.
There would be no point in tracking individual molecules in a bottle of
gas, even if this was practical.
Only a small number of statistical features are relevant.
A substantial part of thermodynamics need not even be based on
statistical physics. Once the interesting quantities are identified,
their mutual relations may not be obvious and they repay study...
This global approach to thermodynamics
starts with the notion of internal energy (U) and/or
with other thermodynamical potentials
(H, F, G ...) measured in energy units.
As the name implies, the variation of the internal energy
(U) of a system is
an accurate account of all forms of energy it exchanges with the rest of the Universe.
In the simplest cases, the variation (dU) in internal energy boils down to the
mechanical work (dW) done to the system and
the quantity of heat (dQ)
which it receives. The first law of thermodynamics
then reads:
dU = dQ + dW
U is a function of the system's state variables,
so its variation is a differential form
of these (as denoted by a straight "d") whereas the same need not be true
of Q and W,
which may depend separately on other external conditions (that's what
the greek d is a reminder of).
A few exchanges of energy can be traced to obvious changes in quantities which are
called
extensive (loosely speaking, a physical quantity is called
extensive when
the measure of the whole is the sum of the measures of the parts).
One example of an extensive quantity is the volume (V) of a system.
A small change in an extensive quantity entails a proportional change in energy.
The coefficient of proportionality is the associated intensive
quantity. The intensive quantity associated to volume is pressure...
dW = - pe dV
That relation comes from the fact that
the mechanical work done by a force is the (scalar) product of
that force by its displacement
(i.e., the infinitesimal motion of the point which yields to it).
In the illustrated case
of a "system" consisting of the blue gas and the red piston,
we must (at the very least) consider the kinetic energy of the piston,
whose speed will change because of the net force which results from
any difference between the external pressure (pe)
and the internal pressure (p).
However, in the very special case of extremely slow changes
(a quasistatic transformation)
the kinetic energy of the piston is utterly negligible
and the internal pressure (p) remains nearly equal to the slowly evolving
external pressure:
dU = - p dV
Now, we can't give a general expression for dU valid for more general
transformations unless
some new extensive variable is involved
(besides volume).
Our intial explanation involving the piston's momentum is certainly valid
(momentum is an extensive variable) but it can't be the "final" one,
since common experience shows that the piston will eventually stop.
The piston's energy and/or momentum must have been "dissipated" into something
commonly called heat.
Could heat itself be the extensive quantity involved in the energy
balance of an infinitesimal energy balance for an irreversible transformation?
The answer is a resounding no.
This misguided explanation would essentially be equivalent to
considering heat as some kind of conserved fluid (formerly dubbed "caloric").
The naive caloric theory was first shown to be untenable
by Rumford in 1798.
The pioneering work of Carnot (1824)
was only reconciled with the first law by Rudolf Clausius
in 1854, as he recognized the importance of the ratio
dQ/T
of the quantity of heat dQ
transferred at a certain temperature T.
In 1865, this ratio was equated to a change dS in the relevant fundamental
extensive quantity for which Clausius himself
coined the word entropy.
Like volume (V) is associated with pressure (p),
entropy (S) is associated with the intensive quantity called
thermodynamical temperature (T) or
"temperature above absolute zero", which can be defined as the reciprocal
of the integrating factor of heat...
This is a linear function of (modern) customary
measurements of temperature. The SI unit
of thermodynamical temperature is the kelvin
(not capitalized).
It's abbreviated K, and should not be used with the word "degree" or the "°" symbol
(unlike the related "degree Celsius"
which refers to a scale originating at the ice point, at 0°C
or 273.15 K, instead of the
absolute zero at 0 K or -273.15°C).
Nowadays, 0°C is exactly equal to
273.15 K and approximately equal to
the ice point (the temperature of melting ice under 1 atm
of pressure). By definition of the Kelvin scale, the temperature of the
triple point of water is exactly 273.16 K
or 0.01°C
[currently].
A system at temperature T
that receives a quantity of heat dQ
(from an external source at temperature Te )
undergoes a variation dS in its entropy:
dS = dQ / T
Conversely, the source "receives" a quantity of heat
- dQ and its own
entropy varies by
- dQ / Te.
The total change in entropy thus entailed is:
dQ ( 1/T
- 1/Te )
The second law of thermodynamic
states that this is always a nonnegative quantity
(total entropy never decreases)...
The system can receive a positive quantity of heat
(dQ > 0)
only from a warmer source (Te > T).
A statistical definition of entropy (Boltzmann's relation)
was first given by Ludwig Boltzmann (1844-1906)
in 1877, using the constant k now named after him.
In 1948, Boltzmann's definition of entropy was properly
generalized by
Claude Shannon (1916-2001)
in the newer context of information theory.
The general expression for infinitesimal transformations (reversible or not)
in the case of an homogeneous gas
(i.e., an hydrostatic system) is simply:
dU = T dS - p dV
In a quasistatic transformation
(p = pe)
the two components of dU can be identified with
heat transferred and work done
( to the system) :
dQ = T dS
dW = - p dV
The above applies to an hydrostatic system involving only
two extensive quantities (entropy and volume) but it generalizes nicely
according to the following pattern which gives the variation (dU) in internal energy
as a sum of the variations of several extensive quantities,
each weighted by an associated intensive quantity:
Extensive:
Entropy (S)
Volume
Area
Length
Electric charge
etc.
dU =
T dS
- p dV
+ a dA
+ f dL
+ j dq
+ ...
Intensive:
Temperature
Pressure
Surface tension
Tensile force
Voltage
For a system whose state at equilibrium is described by N extensive
variables (including entropy)
the right-hand side of the above includes N terms (N=2 for an hydrostatic
system). This equation is the differential version of the relation which gives
internal energy in term of N variables, including entropy.
That same relation may also be viewed as giving entropy in terms of N variables,
including internal energy. A state of equilibrium is characterized by
a maximal value of entropy.
Relativistic Thermodynamics:
Total Hamiltonian Energy (E)
If the system is in (relativistic) motion at velocity v
and momentum p, it may receive some
mechanical workv.dp
which translates into a change of its overall kinetic energy and, thus,
of its total Hamiltonian
energy (E) :
For more details about other equations of relativistic thermodynamics and
the historical controversies about them, see below.
(2005-06-14) 1948:
Claude Shannon's Statistical Entropy (S)
From here to certainty, entropy measures the lack of information.
Consider an uncertain situation described by
W distinct
mutually exclusive
elementary events whose probabilities add up to 1.
If the n-th
such event has probability
pn , then Shannon defined
the statistical entropy S as:
S ( p1 , p2
, ... , pW )
=
W
å
n =1
- k
pn Log (pn )
In this, k is an arbitrary positive constant, which effectively defines
the unit of entropy.
In physics, entropy is normally expressed in joules per kelvin (J/K),
logarithms are natural logarithms, and k is
Boltzmann's constant.
Besides other energy-to-temperature ratios of units,
entropy may also be expressed in
units of information, as discussed below.
Apparently, only two units of entropy have ever been given a specific name but
neither became popular: The boltzmann is
the unit which makes k equal to unity in the above defining relation
(one boltzmann is about
1.38 10-23 J/K).
The clausius is a practical unit best defined as
1000 thermochemical
calories per degree Celsius, namely 4184 J/K (exactly).
In practice, the "clausius" has always been much less popular
than either the cal/K (in the old days)
or the J/K (nowadays).
So defined, the statistical entropy S is nonnegative.
It's minimal (S = 0)
when one of the elementary events is certain.
For a given W, the
entropy S is maximal when every elementary event has the same probability,
in which case
S = k Log (W).
The relation
S = k Log (W)
is known as Boltzmann's Relation.
It's named after Ludwig Boltzmann
(1844-1906) who introduced it in a context
where W was the number of possible states within a
small interval of energy of fixed width, near equilibrium (where all states
are, indeed, nearly equiprobable). As the width of such an interval
is arbitrary, an arbitrary additive quantity was involved
in Boltzmann's original definition (before
quantum theory removed that ambiguity).
Boltzmann's constant
k = R/N
is the ratio of the ideal gas constant (R) to Avogadro's number (N).
Planck named k
after Boltzmann.
According to Misha Gromov
(2014 video)
the above formula was first conceived by Planck, who derived it from
Boltzmann's equiprobable definition (the two men corresponded on the subject).
Claude Elwood Shannon (1916-2001)
made the following key formal remark: Up to a positive factor,
the above definition yields the only nonnegative
continuous function which can be consistently computed by splitting events into
two sets and equating S to the sum of three terms,
namely the two-event entropy for
the split and the two conditional entropies weighted by the respective
probabilities of the splitting sets:
p
=
p1 + p2 + ... + pn
q
=
pn+1 + pn+2 + ... +
pW
= 1- p
S ( p1 ... pW )
=
S (p,q) +
p S (p1 /p ... pn /p) +
q S (pn+1 /q ... pW /q)
Units
used in Computer Science and/or Information Theory
In information theory, the unit of entropy and/or information is the
bit, namely the information
given by a single binary digit (0 or 1).
In the above, this means
k = 1/Log(2). [ k = 1
if logarithms are understood to be in base 2, but "lg(x)" is
a better notation for the binary logarithm of x, following
D.E. Knuth. ]
When it's necessary to avoid the confusion between a binary digit (bit)
and the information it conveys, this unit of information is best called
a shannon (symbol Sh).
The word "bit" itself was coined by the Princeton mathematician
John Tukey (1915-2000)
at Bell Labs around 1950.
Werner Buchholz (IBM)
started calling 8 bits a "byte" in 1956.
Units obtained by multiplying either of those by a
power of 1024 have since become very popular...
Other odd units of information are virtually unused.
For the record, this includes the hartley (symbol Hart)
which is to a decimal digit what the shannon (Sh)
is to a binary digit; the ratio of the former to the latter is:
Therefore, 1 Hart is about 3.322 Sh.
This unit, based on decimal logarithms, was named after
Ralph V.L. Hartley
(1888-1970, of radio oscillator fame)
who introduced information as a physical quantity in 1927-1928
(more than 20 years before Claude Shannon's
Information Theory).
The nat or nit is another unused unit of information
(preferably called a boltzmann as a physical unit of entropy)
which is obtained by letting k = 1 in the above defining equation,
while retaining natural logarithms:
One nat is about 1.44 Sh = 1 boltzmann
(and a nit is about 1.44 bits).
If you must know, a bit (or, rather, a shannon) is about
9.57 10-24 J/K...
A picojoule per kelvin (pJ/K) is about
12 gigabytes (12.1646 GB).
(2005-06-19) The
Third Law: Nernst's Principle (1906)
On the inaccessible state where both entropy and temperature are zero.
The definition of statistical entropy in absolute terms makes it
clear that S would be zero only in some perfectly well-defined pure quantum state.
Other physical definitions of entropy fail to be so specific and leave
open the possibility that an arbitrary constant can be added to S.
The principle of Nernst (sometimes called the "third law" of thermodynamics)
reconciles the two perspectives by stating that entropy must be zero at
zero temperature. Various forms of this law were stated by
Walther Hermann Nernst (1864-1941;
Nobel
1920) between 1906 and 1912.
A consequence of this statement is the fact that nothing can be cooled down to the
absolute zero of temperature (or else, there would be a prior cooling apparatus
with negative temperature and/or entropy).
In the limited context of classical thermodynamics,
the principle of Nernst thus justifies the very existence of the absolute zero,
as a lower limit for thermodynamic temperatures.
Violations of Nernst's Principle :
From a quantum viewpoint, the principle of Nernst would be rigorously true
only if the ground state of every system was nondegenerate
(i.e., if there was always only one quantum state of lowest energy).
Although this is not the case, there are normally very few quantum states of
lowest energy, among many other states whose energy is almost as low.
Therefore, the statistical entropy at zero temperature is always extremely small,
even when it's not strictly equal to zero.
In practice, metastable conditions
present a much more annoying problem:
For example, although crystals have a lower entropy than glasses, some glasses
transform extremely slowly into crystals and may appear absolutely stable...
In such a case, a substance may be observed to retain a significant positive entropy
at a temperature very near the absolute zero of temperature.
This is only an apparent violation of the principle of Nernst.
(2006-09-18) Thermodynamic Potentials Ad hoc substitutes for internal energy or entropy.
Thermodynamic potentials are functions of the state of the system
obtained by subtracting from the internal energy (U) some
products of conjugate quantities (pairs of intensive and extensive quantities,
like -p and V).
They have interesting physical interpretations in common circumstances.
For example, under constant (atmospheric) pressure
enthalpy (H=U+pV)
describes all energy exchanges except mechanical work.
That's why chemists focus on
changes in enthalpy for chemical reactions,
in order to rule out whatever irrelevant mechanical work is exchanged with
the atmosphere in a chemical explosion
(involving a substantial change in V).
As illustrated below, free enthalpy
(Gibbs' function, denoted G) is a convenient way to deal with a
phase transition,
since such a transformation leaves G unchanged,
because it takes place at constant temperature and constant pressure.
More generally, the difference in free enthalpy between two states of equilibrium
is the least amount of useful energy (excluding both
heat and pressure work)
which the system must exchange with the outside to go from one state to the other.
One non-hydrostatic example is a battery of electromotive force e
(i.e., e is the voltage at the electrodes when no current
flows) and internal resistance R
as it delivers a charge q in a time t. The longer the time t, the closer we are
to the quasistatic conditions which make the transfer of energy approach
the lower limit imposed by the change in G, according to the following inequality:
DU =
(VA - VB ) i t =
(Ri - e) q > -e q =
DG
Thermodynamic
Potentials for an Hydrostatic System
The tabulated differential relations are of the following mathematical form:
dz =
(¶z/¶x) dx
+ (¶z/¶y) dy
The matching Maxwell relations in the last-column simply state that
¶2z /
¶y¶x =
¶2z /
¶x¶y
Such statements may be mathematically trivial
(Clairaut, 1740)
but they are quite interesting physically...
For example, from the
equation of state of a gas
(i.e., the relation between its volume, temperature and pressure)
the last two relations give the isothermal derivatives
of entropy with respect to pressure or volume.
This can be integrated to give an expression of entropy involving
parameters which are functions of temperature alone.
(See example below in the
special case of a Van der Waals fluid.)
Thermal capacity is defined as the ratio of the heat received to the
associated increase in temperature.
For an hydrostatic system,
that quantity comes in two flavors:
isobaric (constant pressure) and isochoric
(constant volume) :
Cp = T (
¶S
)p
= (
¶H
)p
¶T
¶T
CV = T (
¶S
)V
= (
¶U
)V
¶T
¶T
The difference between those two happens to be a quantity which can easily
be derived from the
equation of state (the relation linking p, V and T):
By definition, the adiabatic coefficient
is the ratio
g = Cp / Cv
(equal to 1+2/j where
j is 3, 5 or 6 for a classical perfect gas
obeying Joule's law).
The adiabatic coefficient may also be expressed as other ratios of noteworthy
quantities...
(2013-01-15) Relating isothermal
and isentropic derivatives.
The latter must be used to compute the speed of sound.
Sound is a typical isentropic phenomenon
(i.e., for reasonnably small intensities,
sound is reversible and adiabatic).
When quick vibrations are used to probe something, what we're feeling are
isentropic derivatives.
On the other hand, slow measurements at room temperature allow
thermal equilibria with the room at the beginning and the end of the observed transformation.
In that case, we are measuring isothermal coefficients.
Consider, for example, the stiffness K of a fluid or a solid
(more precisely called bulk modulus of elasticity ).
Its inverse is the
relative reduction in volume caused by an increase in pressure.
It comes in two flavors:
Isothermal :
1
= -
1
(
¶V
)T
KT
V
¶p
Adiabatic :
1
= -
1
(
¶V
)S
KS
V
¶p
I'm ignoring the name (compressibility) and the symbol
(k) for the reciprocal of stiffness.
The other two well-established elasticity coefficients
expressed in the same units have unused reciprocals.
We'll need the volumetric thermal expansion coefficient, defined by :
b =
1
(
¶V
)p
V
¶T
Here goes nothing (Maxwell's fourth relation is used in the third line
and the fourth line is obtained by expanding the leading factor of the last term).
dV
=
(
¶V
)S
dp
+ (
¶V
)p
dS
¶p
¶S
(
¶V
)T
=
(
¶V
)S
+ (
¶V
)p
(
¶S
)T
¶p
¶p
¶S
¶p
-V
=
-V
- (
¶V
)p
(
¶V
)p
KT
KS
¶S
¶T
1
=
1
+ (
¶T
)p
(
¶V
)p
b
KT
KS
¶S
¶T
1
=
1
+
T b 2
KT
KS
Cp/ V
The quantity Cp/ V (which appears as the denominator of the
last term) is the molar heat capacity per molar volume; it's also equal
to the mass density r multiplied into
the specific heat capacity cp (note lowercase).
All told, that term is the inverse of a quantity W homogeneous to a pressure,
an elasticity coefficient, or an energy density
(more than 10 years ago,
I proposed the term
thermal wring as a pretext for using the symbol W, which isn't
overloaded in this context):
W =
Cp
=
r cp
=
KS
=
g KT
=
KS KT
V T b 2
T b 2
g - 1
g - 1
KS - KT
(2013-01-17) The thermal Grüneisen parameter z
Adiabatic derivative of Log T with respect to Log 1/V
(or Log r).
Many authors use g
(or gth ) to denote this Grüneisen parameter.
I beg to differ (to prevent confusion with the adiabatic coefficient).
z
= -
V
(
¶T
)S
=
r
(
¶T
)S
T
¶V
T
¶r
z
is the adiabatic ratio of the logarithmic differentials of two quantities:
temperature and either density or volume
(those two differ by sign only).
The relation to the adiabatic coefficient
g = Cp / Cv
= Ks / KT is simply:
g = 1 +
z b T
For condensed states of matter (liquids or solids)
the volumetric coefficient of thermal expansion (b)
is quite small and the above adiabatic coefficient remains very close to unity;
the Grüneisen parameter is more meaningful
(the adiabatic coefficient is traditionally reserved to the study of gases).
This shows that CV is a function of temperature alone.
So, we may as well evaluate it for large molar volumes
(very low pressure) and find that:
CV = j/2 R
That relation comes from the fact that, at very low pressure, the energy of interaction
between molecules is negligible. Therefore, by the
theorem of equipartition of energy,
the entire energy of a gas is the energy which gets equally distributed
among the j active degrees of freedom
of each molecule, including the
3 translational degrees of freedom which are
used to define temperature
and 0, 2 or 3 rotational degrees of freedom
(we assume the temperature is low enough for vibrations modes of the molecules to
have negligible effects; see below).
All told:
j = 3 for a monoatomic gas,
j = 5 for a diatomic gas, j = 6 otherwise.
S = S0 +
j/2 R Log (T) + R Log (V-b)
There's no way to reconcile this expression with Nernst's third law
to make the entropy vanish at zero temperature. That's because the domain of validity
of the Van der Waals equation of state does not extend all the
way down to zero temperature (there would presumably be a transition to a
solid phase at low temperature, which is not accounted for by the model).
So, we may as well accept the classical view, which defines
entropy only up to an additive constant and choose the following expression
(the statistical definition of entropy, ultimately
based on quantum considerations, leaves no such leeway).
Entropy of a Van der Waals fluid ( j = 3, 5 or 6 )
S = R Log [ T j /2 (V-b) ]
Thus, the isentropic equation of such a fluid
generalizes one of the formulations valid for an ideal gas,
when b = 0 and j/2 = 1/(g-1) :
T j /2 (V-b) = constant
Unlike CV, Cp =
g CV
is not constant for a Van der Waals fluid,
since:
Cp -
CV
=
T (
¶p
)V
(
¶V
)p
¶T
¶T
(2012-07-03) The
empirical Dulong-Petit law (1819) :
The heat capacity per mole is nearly the same
(3R) for all crystals.
In 1819, Dulong and Petit (respectively the third and the second
holder of the chair of physics at Polytechnique
in Paris, France) jointly observed that the heat capacity of metallic
crystals is essentially proportional to their number of atoms.
They found it to be nearly 25 J/K/mol for every solid metal
they investigated (this would have failed at very low temperatures).
In 1907, Albert Einstein gave a
simplified quantum model,
which explained qualitatively why the Dulong-Petit law fails at low temperature.
His model also linked the empirical values of the molar heat capacity
(at high temperatures) to the ideal gas constant R :
In 1912, Peter Debye devised an even
better model
(equating the solid's vibrational modes with propagating phonons )
which is also good at low temperatures.
Its limited accuracy at intermediate temperatures is entirely due to the simplifying assumption
that all phonons travel at the same speed.
When applied to a gas of photons, that statement is true
and the model then describes blackbody radiation perfectly,
explaining Planck's law !
wangshu
(2010-12-19) Vibrational modes of the
CO2 molecule.
What do the following energy levels contribute to the heat
capacity of carbon dioxide at 400 K?
En = (n+1) hn
where n = 20 THz
The ratio
hn / kT = 2.4
indicates that the quanta involved are similar to the average energy
of a thermal photon
(2.7 kT).
(2005-06-25) Latent Heat (L) and Clapeyron's Relation
As entropy varies in a change of phase,
some heat must be transferred.
The British chemist
Joseph
Black (1728-1799) is credited with the
1754 discovery of fixed air (carbon dioxide) which helped
disprove the erroneous phlogiston theory of combustion.
James Watt (1736-1819)
was once his pupil and his assistant.
Around 1761, Black observed that a phase transition
(e.g., from solid to liquid) must be accompanied by a transfer of
heat, which is now called latent heat.
In 1764, he first measured the latent heat of steam.
The latent heat L is best described as the difference
DH
in the enthalpy (H=U+pV) of the two phases, which accurately
represents heat transferred under constant pressure
(as this voids the second term in
dH = TdS + Vdp).
Under constant pressure, phase transistion
occurs at constant temperature.
So, the free enthalpy
(G=H-TS) remains constant
(as dG = -SdT + Vdp).
Consider now how this free enthalpy G varies along
the curve which gives the pressure p
as a function of the temperature T
when the two phases 1 and 2 coexist.
Since G is the same on either side of this curve, we have:
dG = -S1 dT + V1 dp
dG = -S2 dT + V2 dp
Therefore, dp/dT is the ratio
DS/DV
of the change in entropy to the change in volume
entailed by the phase transition.
Since TDS = DH, we obtain:
The Clausius-Clapeyron Relation :
T dp/dT =
DH / DV
= L / DV
That relation is one of the nicest results
of classical thermodynamics.
(2020-01-17) Phase Diagram of Carbon
Either diamond or graphite can be metastable in the domain of the other.
The boundary between the domains of graphite and diamond is
well-known:
In the plot of Log(p) as a fubction of T,
tt's a straight line between the point at 0 K of temperature and 1.7 GPa
of pressure and the diamond-graphite-liquid triple point
(5000 K, 12 GPa).
Meeting at that triple-pont on either side or that boundary are two other straight lines
between which one carbon allotrope is metastable in the domain where the other
is stable. Thus, diamond is ordinarily metastable but would
morph into graphite at high temperature and low pressure.
(2006-09-23) Joule-Thomson Coefficient and
Inversion Temperature.
A flow expansion of a real gas may cool it enough to liquefy it.
Joule Expansion & Inner Pressure
Expanding dS along dT and dV, the expression dU = T dS - p dV becomes:
dU
=
T (
¶S
)V
dT
+ [
T (
¶S
)T
-
p ] dV
¶T
¶V
=
CV
dT
+ [
T (
¶p
)V
-
p ] dV
¶T
This gives the following expression (vanishing for a perfect gas) of the
so-called Joule coefficient which tells
how the temperature of a fluid varies when it undergoes a
Joule expansion, where the internal energy (U) remains constant.
An example of a Joule expansion is the removal of a separation between the gas
and an empty chamber.
(
¶T
)U
= -
1
[
T (
¶p
)V
-
p ]
¶V
Cv
¶T
The above square bracket is often called the
internal pressure or
inner pressure.
It's normally a positive quantity which repays study.
Let's see what it amounts to in the case of a
Van der Waals fluid;
(
¶U
)T
=
[
T (
¶p
)T
-
p ] =
RT
- p =
a
¶V
¶T
V-b
V2
By integration, this yields:
U = U0(T) - a / V.
The latent heat of liquefaction (L) is obtained in term of the
molar volumes of the gaseous and liquid phases (VG,VL) either as
DH = DU+pDV or as TDS
(using the above expression for S):
L =
p (VG-VL) + a (1/VL-1/VG)
= RT Log [ (VG-b) / (VL-b) ]
Joule-Thomson (Joule-Kelvin) Expansion Flow Process
The Joule-Kelvin
coefficient
(m) pertains to an isenthalpic expansion.
Its value is obtained as above (from an expression of dH instead of dU):
m =
(
¶T
)H
=
1
[
T (
¶V
)p
-
V ]
=
V
( T a - 1 )
¶p
Cp
¶T
Cp
m vanishes for perfect gases
but allows an expansion flow process which can cool many real gases
enough to liquefy them, if the initial temperature is below the
so-called inversion temperature, which makes
m positive.
More precisely, the inversion temperature is a function of pressure.
In the (T,p) diagram, there is a domain where isenthalpic decompression causes cooling.
The boundary of that domain is called the inversion curve.
In the example of a
Van der Waals fluid, the equation of the inversion curve is
obtained as follows:
0 = T(
¶V
)p
- V =
R T
- V
¶T
p -
a / V2 +
2 a b / V3
This gives a relation which we may write next to the equation of state:
R T
=
p V -
a / V + 2 a b / V2
R T + p b
=
p V +
a / V - a b / V2
By eliminating V between those two equations, we obtain a single relation
which is best expressed in units of the critical point (pc = 1,
Tc = 1):
T±
=
15 / 4 + p / 24 ±
Ö
9 - p
If T is above T+
(or below T- )
then decompression won't cool the gas.
At fairly low pressures, the inversion temperature is approximately:
Ti = 6.75 Tc
The ratio observed for most actual gases is lower than 6.75:
Although it's 7.7 for helium, it's only
6.1 for hydrogen,
5.2 for neon,
4.9 for oxygen or nitrogen, and
4.8 for argon.
A Joule-Thomson cryogenic apparatus has no moving parts at low temperature
(note that the cold but unliquefied part of the gas is returned in thermal contact with
the high-pressure intake gas, to pre-cool it before the expansion valve).
The effect was described in 1852 by William Thomson (before he became
Lord Kelvin).
So was the basic design, with the cooling countercurrent.
Several such cryogenic devices can be "cascaded" so that
one liquefied gas is used to lower the intake temperature of the next apparatus...
Liquid oxygen was obtained this way in 1877,
by Louis Paul Cailletet (France)
and Raoul Pierre Pictet (Switzerland).
Hydrogen was first liquefied in 1898, by Sir James Dewar (1842-1923).
Finally, helium was liquefied in 1908, by the Dutch physicist
Heike Kamerlingh Onnes (1853-1926; Nobel 1913).
In 1895, the German engineer
Carl von Linde (1842-1934)
designed an
air liquefaction machine based on this throttling process,
which is now named after him (Linde's method).
The direct conversion of heat into electricity at the junction of two different metals was
discovered in 1794 by Alessandro Volta (1745-1827).
This was rediscovered in 1821 by
Thomas Johann Seebeck
(1770-1830) who observed only the ensuing deflection of a nearby compass needle
and termed the effect thermomagnetic.
Ørsted (who had discovered only a few months
earlier that an electric current has magnetic effects) realized that
an electric current was the proper cause and he wisely renamed the
effect thermoelectric, which now stands.
The effect happens to be thermodynamically reversible
so that if two junctions of unlike metals are connected in a circuit,
then one of the is heated and the other is cooled
(according to the direction of the current).
The ensuing possibility of direct electric cooling was discovered in 1834
by the French physicist Jean-Charles Peltier and the phenomenon
is now called Peltier effect in his honor.
Now, it should be clear from its statistical definition
that entropy is a relativistic invariant,
since the probability of a well-defined spacetime event cannot depend on the
speed of whoever observes it.
Mercifully, all reputable authors agree on this one...
They haven't always agreed on the following (correct)
formula for the temperature T
of a body moving at speed v whose temperature is
T0 in its rest frame :
Mosengeil's Formula (1906)
T =
T0
Ö
1 -
v2/c2
The invariance of the entropy S
means that a quantity of heat
(dQ = T dS)
transforms like the temperature T.
So do all the thermodynamic potentials,
including internal energy (U)
free energy (F = U-TS)
Helmholtz' enthalpy (H = U+pV)
and Gibbs' free enthalpy (G = H-TS)...
dQ
=
dQ0
Ö
1 -
v2/c2
This was wrongly ignored by
Eddington
(and several later authors) who assumed
that heat and thermodynamical potentials ought to transform
like mechanical energy, because they are measured in the same units.
In 1952, Einstein himself
recanted
his 1907 support of the above...
Such bizarre episodes are discussed at length in the
1967 dissertation of Abdelmalek Guessous,
under Louis de Broglie.
Note that the ideal gas law (pV = RT)
is invariant because the pressure p is invariant, whereas the temperature T
transforms like the volume V.
(The same remark would hold for any gas obeying Joule's second law.)
One of several ways to justify the above expression for the temperature of a moving
body is to remark that the frequency of a typical photon from a moving
blackbody is proportional to its temperature.
Thus, if it can be defined at all, temperature must
transform like a frequency.
This viewpoint was first expounded in 1906 by
Kurd von Mosengeil
and adopted at once by Planck,
Hasenöhrl and
Einstein
(who would feel a misguided urge to recant, 45 years later).
In 1911, Ferencz Jüttner (1878-1958) retrieved the same formula for a moving gas,
using a relativistic variant of an earlier argument of
Helmholtz. He derived the
relativistic speed
distribution function recently confirmed numerically
(2008-04-23)
by Constantin Rasinariu in the case of a 2-dimensional gas.
(In his 1964 paper
entitled Wave-mechanical approach to relativistic thermodynamics,
L. Gold gave a quantum version of Jüttner's argument.)
Mosengeil's (correct) formula was also featured in the textbook published by
Max von Laue in 1924.
By that time,
Eddington had already published his own 1923 textbook, containing
the aforementioned erroneous idea which
other people would later have independently
(apparently, everybody overlooked that part of Eddington's famous book
until A. Gamba quoted it in 1965).
The ensuing mess is still with us.
(
2012-03-10
)
In 1967, under the supervision of
Louis de Broglie,
Abdelmalek Guessous completed a full-blown attack,
using Boltzmann's statistical mechanics
(reviewed below).
This left no doubt that thermodynamical
temperature must indeed transform as stated above
(in modern physics, other flavors of temperature are not welcome).
Equating heat with a form of energy was once a
major breakthrough, but the
fundamental relativistic distinction
between heat and Hamiltonian energy
noted by most pioneers (including Einstein in his youth)
was butchered by others (including Einstein in his old age)
before its ultimate vindication...
A few introductions to Relativistic Thermodynamics :
Richard Chace Tolman (1881-1948)
"Relativity, Thermodynamics, and Cosmology" (1934)
Oxford University Press (Dover 1987 Reprint: ISBN 0-486-65383-8)
Louis de Broglie (1892-1987;
Nobel 1929)
"La Thermodynamique de la particule isolée" (1964)
Gauthier-Villars, Paris 1964.
Abdelmalek Guessous
[Amazon
| Google]
305 pages (93 from thesis)
"Thermodynamique relativiste"
Gauthier-Villars, Paris 1970.
That book (reviewed below) is dear to me:
I bought a used copy
(for 98 FF) with only 2 other books,
on a fateful trip to Paris after high-school graduation, in the Summer of 1973,
just before entering Taupe Laplace.
The mature selection I made on a tight budget amazes me now.
The other books were:
Philosophie mathématique (1962) by
Jean Cavaillès
(on Set Theory) and
Distributions et transformation
de Fourier (1971) by François Roddier (Fourier transform).
In 1968,
Pierre-V.
Grosjean
called the waves of controversies about Mosengeil's formula
the temperature quarrel...
In spite or because of its long history, that dispute is regularly revived
by authors who keep discarding one fundamental subtlety of thermodynamics:
Heat doesn't
transform like a Hamiltonian energy
(which is the time-component of an energy-momentum 4-vector) but
like a Lagrangian.
Many essays are thus
going astray, including:
I stand firmly by the statement that if
temperature can be defined at all, it must obey
Mosengeil's formula.
The following articles argue against the premise of that conditional
proposition, at least for one type of thermometer:
On April 14, 1967,
Guessous defended his doctoral dissertation (Recherches sur la thermodynamique relativiste).
He established the relativistic thermodynamical temperature
to be the reciprocal of the integrating factor of the quantity of heat,
using the above statistical definition of entropy.
This superior definition is shown, indeed, to be
compatible with Mosengeil's formula.
The actual text isn't easy to skip through, because the author keeps waving the formulas he
is arguing against
(mainly the Ott-Arzeliès formulation, inaugurated by Eddington and
ruled out experimentally by P. Ammiraju in
1984).
In 1970, Guessous published an expanded version of the thesis as a
book entitled "Thermodynamique relativiste",
prefaced by his famous adviser
Louis de Broglie (Nobel 1929).
That work is still quoted by some scholars,
like Simon Lévesque,
from Cégep
de Trois-Rivières
(2007-01-13)
although the subject is no longer fashionable.
The original dissertation of Abdelmalek Guessous appears verbatim as
the first five chapters of the book (93 out of 305 pages).
Unfortunately, Guessous avoids contradicting (formally, at least)
what was established by his adviser for pointlike systems.
This impairs some of the gems found in Chapter VI and beyond,
because the author retains his early notations even as he shows them to be dubious.
For example, the paramount definition of internal energy
as a thermodynamical potential (transforming like a Lagrangian)
doesn't appear until Chapter VI where it's dubbed U',
since U was used throughout the thesis for
the Hamiltonian energy E (not clearly identified as such).
More importantly, Guessous runs into the correct definition
of the inertial mass (namely, the momentum to velocity ratio)
but keeps calling it "renormalized mass"
(denoted M' ) while awkwardly retaining the symbol
M as a mere name for E/c2
(denoted U/c2 by him)
which downplays the importance of the aforementioned true inertial mass
m = M'. So, Guessous
missed (albeit barely so) the revolutionary expression of the
inertia of energy for N interacting particles at a nonzero
temperature T, presented next
in the full glory of traditional notations consistent with the rest of this page.
(2008-10-07) Inertia of Energy at Nonzero Temperature
The Hamiltonian energy
E is not proportional to the inertial mass M.
Here's the great formula which I obtained many
years ago by pushing to their logical
conclusion some of the arguments presented in the 1969 addenda to the
1967 doctoral dissertation of Abdelmalek Guessous.
I've kept this result of my younger self in pectore
for far too long
(I first read Guessous' work in 1973).
E = M c 2
- N k T
We define the inertial mass (M) of a relativistic
system of N
point-masses as the ratio of its total momentum p
to the velocity v of its center-of-mass:
p = M v
It's not obvious that the
dynamical momentum p is actually
proportional to the velocity v so that
M turns out to be simply a scalar quantity !
The description of a moving object of nonzero size always takes place
at constant time in the frame K of the observer.
The events which are part of such a description are simultaneous in K
but are usually not simultaneous in the rest frame
(Ko ) of the object.
That viewpoint has been a basic tenet of Special
Relativity ever since Einstein showed in excruciating details
how it explains the
Lorentz-Fitzgerald contraction,
which makes the volume V of a moving solid
appear smaller than its volume at rest V0 :
V
=
V0
Ö
1 -
v2/c2
(2008-10-13) Angular Momentum
Local temperature is higher on the outer parts of a rotating body.
(2005-06-20) Stefan's Law. The Stefan-Boltzmann Law.
Each unit of area at the surface of a black body radiates a total power
proportional to the fourth power of its thermodynamic temperature.
Jozef Stefan
This law was discovered experimentally in 1879, by the Slovene-Austrian physicist
Joseph (Jozef) Stefan (1835-1893).
It would be justified theoretically in 1884, by Stefan's most famous student:
Ludwig Boltzmann (1844-1906).
The energy density (in J/m3 or Pa) of the thermal
radiation inside an oven of thermodynamic temperature T (in K) is
given by the following relation:
[ 4 s / c ] T 4
=
[ 7.566 10-16 Pa/K4 ] T 4
On the other hand, each unit of area at the surface of a black body
radiates away a power proportional to the fourth power of its temperature T.
The coefficient of proportionality is
Stefan's constant
(which is also known as the Stefan-Boltzmann constant). Namely:
s =
( 2 p 5 k 4 ) /
( 15 h 3 c 3 )
=
5.6704 10-8 W/m2/K4
Those two statements are related.
The latter can be derived from the former using the following argument,
based on geometrical optics, which merely assumes that
radiation escapes at a speed equal to Einstein's
constant (c).
One of the best physical approximations to an element of the surface
of a "black" body is a small opening in the wall of a large cavity ("oven").
Indeed, any light entering such an opening will never be reflected directly.
Whatever comes in is "absorbed", whatever comes out bears no relation whatsoever
to any feature of what was recently absorbed...
The thing is black in this precise physical sense.
Luminosity of a Spherical Blackbody
L = 4 p s T4 R2
To a good enough approximation, this formula relates the surface temperature
T of a star of radiant absolute luminosity L
to its radius R.
The largest stellar radius so measured was that
of KY Cygni
but this came from an overestimated luminosity due to an artifact of the
reddening correction factor in K-band measurements
(KY Cygni is only one of the largest
known star, not the largest one).
(2005-06-19) A Putative "Fourth Law"
about Maximal Temperature
Several arguments would place an upper bound on temperature...
Several arguments have been proposed which would put a theoretical maximum
to the thermodynamic temperature scale.
This has been [abusively] touted as a "fourth law" of thermodynamics.
Some arguments are obsolete, others are still debated within the latest context
of the standard model of particle physics:
In 1973, D.C. Kelly argued that no temperature could ever exceed a limit
of a trillion kelvins or so, because when particles are heated up, very high
kinetic energies will be used to create new particle-antiparticle pairs rather
than further contribute to an increase in the velocities of existing particles.
Thus, adding energy will increase the total number of particles rather than the
temperature.
This quantum argument is predated by a semi-classical guess,
featuring a rough quantitative agreement:
In 1952, French physicist Yves Rocard
(father of Michel Rocard, who was France's prime minister from 1988 to 1991)
had argued that the density of electromagnetic energy ought not to exceed by much
its value at the surface of a "classical electron"
(a uniformly charged sphere with a radius of about 2.81794 fm).
Stefan's law would then imply an upper limit for temperature
on the order of what has since been dubbed "Rocard's temperature", namely:
3.4423 1010 K
One process seems capable of generating temperatures
well above Rocard's temperature: the explosion
of a black hole via Hawking radiation.
Rocard's temperature would be that of a black hole of about
8 1011 kg, which is much too small
to be created by the gravitational collapse of a star.
Such a black hole could only be a "primordial" black hole,
resulting from the hypothetical
collapse of "original" irregularities
(shortly after the big bang).
Yet, the discussion below shows that a black hole whose temperature is
Rocard's temperature would radiate away its energy for a very long time:
about 64 million times the age of the present Universe...
It gets hotter as its gets smaller and older.
As the derivation we gave for Stefan's Law
was based on geometrical optics,
it does not apply in the immediate vicinity
of a black hole (space curvature is important
and wavelengths need not be much shorter than the sizes involved).
A careful analysis would show that a
Schwarzschild black hole absorbs
photons as if it was a massless black sphere (around which space is "flat") with a
radius equal to
a = 3Ö3 MG/c2 (about 2.6 times the Schwarzschild radius).
Thus, it emits like a black body of that size
(obeying Stefan's law). Its power output is:
As this entails a mass loss
inversely proportional to the square of the mass,
the cube of the black hole's mass decreases at a constant rate
of 27 / 20480p
(in natural units).
The black hole will thus evaporate completely after a time proportional
to the cube of its mass, the coefficient of proportionality being about
5.96 10-11 s per cubic kilogram.
A black hole of 2 million tonnes (2 109 kg)
would therefore have a lifetime about equal to
the age of the Universe (15 billion years).
Hawking thus showed that his first hunch was not quite right:
a black hole's area may decrease steadily
because of the radiation which does carry entropy away.
The only absolute law is that, in this process like in any other,
the total entropy of the Universe can only increase.
There is little doubt that Hawking's computations are valid
down to masses as small as a fraction of a gram.
However, it must be invalid
for masses of the order of the Planck mass (about 0.02 mg),
as the computed temperature would otherwise be such that a "typical" photon
would carry away an energy kT equivalent to the black hole's total energy.
This allows the possibility of temperatures more than 15 orders of magnitudes
higher than Rocard's temperature.
The "natural" unit of temperature is 21 orders of magnitude
above Rocard's temperature.
In his wonderful 1977 book The First Three Minutes, 1979 Nobel laureate
Steven Weinberg
gives credit to R. Hagedorn, of the CERN laboratory in Geneva,
for coming up with the idea of a maximum temperature in particle physics when an
unlimited number of hadron species is allowed.
Weinberg quotes work on the subject
by a number of theorists including Kerson Huang (of MIT) and himself, and states
the "surprisingly low" maximum temperature of
2 000 000 000 000 K
for the limit based on this idea...
However, in an afterword to the 1993 edition of his book,
Weinberg points out that the "asymptotically free" theory of strong
interactions made the idea obsolete:
a much hotter Universe would "simply"
behave as a gas of quarks, leptons, and photons (until unknown territory is found
in the vicinity of Planck's temperature).
In spite of these and other difficulties, there may be a maximum temperature
well below Planck's temperature which is not attainable by any means, including
black hole explosions:
One guess is that newly created particles could form a hot shell around
the black hole which could radiate energy back into the black hole.
The black hole would thus lose energy at a lesser rate,
and would appear cooler and/or larger to a distant observer.
The "fourth law" is not dead yet...
(2005-06-20) Hawking Radiation and the Entropy
of Black Holes
Like all perfect absorbers, black holes radiate with blackbody spectra.
The much-celebrated story of this fundamental discovery
starts with the original remark by
Stephen W. Hawking
(in November 1970) that
the surface area of a black hole can never decrease.
This law suggested that surface area is to a black hole what
entropy is to any other physical object.
Jacob Bekenstein (1947-2015)
was then a postgraduate student working at Princeton under John Wheeler.
He was the first to take this physical analogy seriously, before all the
mathematical evidence was in.
Following Wheeler, Bekenstein remarked that black holes swallow the entropy of
whatever falls into them.
If the second law of thermodynamics is to hold
in a Universe containing black holes, some entropy must
be assigned to black holes.
Bekenstein suggested that the entropy of a black hole was, in fact,
proportional to its surface area...
At first, Hawking was upset by Bekenstein's "misuse" of his discovery,
because it seemed obvious that anything having an entropy would
also have a temperature, and that anything having a temperature would radiate away
some of its energy.
Since black holes were thought to be unable to let anything
escape (including radiation) they could not have a temperature or an
entropy. Or so it seemed for a while...
However, in 1973, Hawking himself made acclaimed calculations confirming Bekenstein's hunch:
The entropy (S) of a black hole is proportional to
the area (A) of its event horizon.
S =
k 2p c 3
( ¼ A )
h G
What Stephen Hawking discovered is that quantum effects allow black
holes to radiate (and force them to do so).
One of several
explanatory pictures is based on the steady creations and anihilations of
particle/antiparticle pairs in the vacuum, close to a black hole...
Occasionally, a newly-born particle falls into the black hole before recombining
with its sister, which then flies away as if it had been
directly emitted by the black hole.
The work spent
in separating the particle from its antiparticle comes
from the black hole itself, which thus lost an energy equal to the mass-energy of
the emitted particle.
For a massive enough black hole, Stephen Hawking found the corresponding radiation spectrum
to be that of a perfect blackbody having a temperature proportional to the
surface gravity g of the black hole
(which is constant over the entire horizon of a
stationary black hole).
In 1976, Bill Unruh
generalized this proportionality between any gravitional field g
(or acceleration) and the temperature T of an associated heat bath.
Temperature T is proportional to (surface)
gravity g.
kT
=
g h / (4p2c)
[ Any coherent units ]
T
=
g / 2p
[ In natural units ]
In the case of the simplest black hole
(established by Karl Schwarzschild as early as 1915)
g is c2/2R, where R
is the Schwarzschild radius equal to
2MG/c2
for a black hole of mass M.
In SI units, kT is about 1.694 / M.
(2005-06-13) Statistical Approach :
The Partition Function (Z).
Z is a sum over all possible quantum states: Z(b) =
å exp (-b E)
E is the energy of each state.
b = 1/kT is related to temperature.
A simplified didactic model: N independent spins
Consider a large number (N) of
paramagnetic ions whose locations in a crystal
are sufficiently distant so interactions between them can be neglected.
The whole thing is submitted to a uniform magnetic induction B.
Using the simplifying assumption that each ion behaves like an electron,
its magnetic moment can only be measured to be aligned with the external field
or directly opposite to it...
Thus, each eigenvalue E of the [magnetic] energy of the ions can be expressed in
terms of N two-valued quantum numbers
si = ±1
E = m B
N
å
i =1
si
m is a constant
(it would be equal to Bohr's magneton for electrons).
As the partition function introduced above involves exponentials of such sums, which
are products of elementary factors, the entire sum boils down to:
Z(b)
=
å exp (-b E)
=
[ exp (b m B)
+ exp (-b m B) ] N


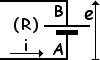 One non-hydrostatic example is a battery of electromotive force e
(i.e., e is the voltage at the electrodes when no current
flows) and internal resistance R
as it delivers a charge q in a time t. The longer the time t, the closer we are
to the quasistatic conditions which make the transfer of energy approach
the lower limit imposed by the change in G, according to the following inequality:
One non-hydrostatic example is a battery of electromotive force e
(i.e., e is the voltage at the electrodes when no current
flows) and internal resistance R
as it delivers a charge q in a time t. The longer the time t, the closer we are
to the quasistatic conditions which make the transfer of energy approach
the lower limit imposed by the change in G, according to the following inequality: