(2017-11-20) Vacuum and the origin of atmospheric pressure.
In still air, the vertical gradient of pressure is the weight density.
At the urging of Blaise Pascal (1623-1662),
on 19 September 1648, Florin Périer (Pascal's brother-in-law) led a party of
several Clermont-Ferrand notables on an ascension
of nearby Puy-de-Dôme
to record the variation of air pressure with altitude,
using a mercury barometer (invented by Torricelli in 1643).
The variation they found was so large that Pascal realized he could observe the effect locally in Paris,
using the modest variation in altitude at the bottom and at the top of the freshly rebuilt
Saint-Jacques Tower
(the tallest building in Paris at the time).
A dozen years later,
on 27 April 1661, Richard Towneley (1629-1707)
and Henri Power (1623-1668; FRS 1663)
farmously performed a similar experiment to measure the pressure of air at different altitudes on
Pendle Hill in Lancashire.
(2006-01-14) The Magdeburg Hemispheres : Mighty Pressure.
A famous demonstration by the inventor of the vacuum pump.
The inventor of the air pump (1650) was the German physicist Otto von Guericke
(1602-1686) who was Mayor of Magdeburg.
Guericke demonstrated the might of atmospheric pressure,
by building two copper bowls, 14 inches in diameter
(the Magdeburg hemispheres) which could form an airtight sphere
from which air could be pumped out.
In 1654, the device was demonstrated dramatically before the Imperial Diet at
Regensburg, in the presence of
Emperor Ferdinand III:
After Guericke had pumped (most of) the air out of that Magdeburg sphere,
two teams of horses couldn't pull apart those two hemispheres which
only the "thin" surrounding air was holding together.
The force holding evacuated 14" Magdeburg hemispheres together is
the product of the atmospheric pressure
(about 101325 Pa) into the cross-sectional area
(a disk 14" across has an area of about
0.1 m2 ). ;
ton of thrust...
(2003-05-21) p V = n R T
Who formulated the ideal gas law ?
However,
Joseph Gay-Lussac (1778-1850)
had first put all aspects of this relation together around 1802.
Although Gay-Lussac only considered a fixed amount of gas (constant n)
at that time, he would also pave the way to Avogadro's law
with his 1809 observation that chemical gaseous reactions always entail
simple ratios in the volumes of the reactants and products.
In 1805, he had famously made that key observation for the formation of
water from oxygen and hydrogen (working with
Alexander von Humboldt).
The ideal gas law
(French: Loi des gaz parfaits) is deceptively simple:
p V = n R T
This states that the product of the pressure (p)
and volume (V) of an ideal gas is proportional to its absolute
temperature (T) and to the number of molecules in it; "n" is actually the number
of moles, which is the number of molecules divided by a numerical
constant called Avogadro's number,
whereas "R" is a constant of proportionality equal to
Boltzmann's constant (k) multiplied into
Avogadro's number:
The law was instrumental in the clarification of all the concepts involved,
with the sole exception of "volume", of course...
The law can be broken down into four statements which were historically
studied separately (some, or all, of these may be invalid
in more elaborate models of actual fluids).
When the full law was finally formulated as above,
it became possible to quantify the discrepancies between the
"ideal" behavior it described and the actual properties of real gases...
Avogadro's Law (1811)
At the same temperature and [low] pressure,
equal volumes of different gases contain the same number of molecules.
This was formulated by Amedeo Avogadro (1776-1856) in 1811.
Like other ideal gas laws, it's only a rough approximation
under most actual conditions. However, it's so accurate at very low pressures
that it can be presented as a limiting case for real gases at constant temperature:
n R T =
lim
p V(p)
p ® 0
Boyle-Mariotte Law : Isothermal (1662, 1676)
At a given temperature, the volume of a gas is inversely proportional to its pressure.
The law was first formulated in print by
Robert Boyle (1627-1691) in 1662.
Boyle omitted the important isothermal requirement which was added in 1676 by
Edme Mariotte (1620-1684)
when he rediscovered the whole thing independently...
A fluid which obeys the Boyle-Mariotte law is called a
Mariotte gas.
In such a gas, the product pV depends only on the absolute temperature (T) :
p V = n R f (T)
Historically, it's essentially the simplification entailed by putting
f (T) = T in the (idealized) above equation
which helped define the concept of "absolute" temperature
on which other aspects of the ideal law depend quantitatively:
Charles' Law : Isobaric (1787)
At a given pressure, the volume of a gas is proportional
to its absolute temperature.
V f (p) = n R T
The isobaric law has been named after
Jacques [Alexandre César]
Charles (1746-1823)
to acknowledge the unpublished work he performed in 1787
(which was quoted by Gay-Lussac in 1802).
Gay-Lussac's Law : Isochoric (1802)
When confined to a given volume, the pressure of a gas is
proportional to its absolute temperature.
p f (V) = n R T
This is the law of perfect gases under isochoric (or isovolumic) conditions.
(2006-09-18) Joule's
Law (Joule's first law) The
internal energy (U) of an ideal gas depends only on its temperature.
This law goes beyond the descriptive aspects of a gas (volume, pressure and temperature)
by introducing energetic aspects:
The internal energy (U) of a gas is
[ideally] a function
of its absolute temperature only.
Skip the following "advanced" discussion on first reading.
Thus, at constant volume, Joule's first law translates into
a differential equation which etablishes that
p and T are proportional.
It is equivalent to the
isochoric law of Gay-Lussac,
which is true if and only if the molar equation of state is of the form:
p f (V) = R T
For a perfect gas, the internal energy is actually proportional to the
absolute temperature, under the classical assumptions of
the kinetic theory of gases,
which defines temperature as proportional to the average (translational) kinetic energy
of molecules and postulates equipartition of energy between the
j active mechanical degrees of freedom of each molecule:
Along with the 3 translational degrees,
the number of rotational degrees of freedom is:
0 for a spherical molecule
(monoatomic gases: helium, argon, etc.).
2 for a molecule with axial symmetry
(carbon dioxide
and all diatomic gases, like hydrogen, nitrogen and oxygen).
The reason why a quantum object can't rotate about
an axis of perfect symmetry is that such a rotation would be
"logically"
impossible to observe, as it does not entail any change in the quantum state.
At high temperature, molecules can no longer be considered perfectly rigid.
Technically, vibrational modes exist in molecules of two atoms
or more. Those behave as additional degrees of freedom that are
"dormant" at low temperatures and
become :active" at higher temperatures (for reasons explained
by statistical mechanics).
If the assumption is made (which is part of the ideal gas model) that there's
no energy of interaction between molecules
(which thus interact only via relatively rare collisions)
the above tells the whole story:
U = (j/2) nRT
U = 3/2 nRT for monoatomic gases
(e.g., He, Ar).
U = 5/2 nRT for diatomic
or linear molecules
(e.g., H2, N2, CO2).
U = 3 nRT for all other gases
(e.g., H2O, NH3, CH4).
Molecular interactions in real gases
entail some violation of Joule's law.
Even nonpolar neighboring molecules induce distortions in each other's
electronic clouds, causing attractive forces called Van der Waals forces.
Joule-Thomson law (Joule's second law):
Joule's second law states that the
enthalpy (H = U+pV) of a
Joule-Thomson gas depends only on its temperature.
The molar equation of state of a Joule-Thomson gas is of the following form,
which is equivalent to the isobaric law of Charles:
V f (p) = R T
A gas which obeys both of Joule's laws is necessarily a
perfect gas.
(2010-12-13) Deflating
a Tire What happens when a pressurized gas is released into the atmosphere?
When air is let out of a tire, the temperature of the tire does not change.
This non-intuitive fact holds for ideal gases
but not necessarily for real gases.
The reason is that each molecule contributes equally to the total pressure,
independently of the other molecules which may or may not be there.
Each molecule that escapes thus reduces the pressure (p) and the number of moles
inside the container (n) in the same proportion. This does not entail
any change in the temperature T = (p/n) V/R.
(2006-09-17) Realistic Equations of State [molar]
Better approximations for actual gases than the ideal gas law.
The equations below are substitutes for the molar (n = 1)
ideal gas law.
They were designed to better describe the actual behavior of real gases.
In particular, the volume of a gas under extreme pressure does not
vanish but tends to a finite limit, called covolume.
The molar covolume is traditionally denoted by the symbol "b".
The simplest (molar) equation which accounts for this important feature of real gases
has been known as the Clausius equation of state :
p ( V-b ) = RT
The Clausius equation for n moles of gas would be:
p ( V-nb ) = nRT.
A Clausius gas thus obeys the isochoric
law of Gay-Lussac and does not really contradict
Avogadro's law (since, at very low pressure,
the covolume of any gas is a negligible fraction of its volume).
On the other hand, a Clausius gas does violate
the isothermal law of Boyle-Mariotte
and the isobaric law of Charles.
In the main, this equation accounts for interactions between molecules
which are able to trigger a transition between liquid and gaseous phases below a certain
temperature, which was apparently first identified as critical
by the French physicist
Charles Cagniard
(1777-1859; X1794) in 1822.
The Irish physicist
Thomas
Andrews (1813-1895) is often credited for the principle of the
"continuity of states" in a fluid: The gaseous and liquid forms
can be transformed into each other without undergoing a phase
transition (by traveling above the critical point).
Andrews showed this in 1869 for carbon dioxide
(which doesn't exist as a liquid below its surprisingly high triple-point
pressure of 5.11 atm).
At the critical temperature,
the isothermal curve which gives pressure as a function of volume has an
inflexion point with an horizontal tangent:
(
¶p
)T
= 0
(
¶2p
)T,T
= 0
¶V
¶V2
Differentiating the equation of state of a Van der Waals fluid, we obtain:
( V-b ) dp + ( p -
a / V2 +
2 a b / V3 ) dV
= R dT
Along an isothermal curve (dT = 0) this yields:
(
¶p
)T
= -
p -
a / V2 +
2 a b / V3
¶V
V - b
At the critical point, that quantity vanishes (the tangent to the isothermal
is horizontal). Since p is thereby stationary at that point,
the isothermal derivative of this expression is simply equal to its derivative
with respect to V. The vanishing of that
second derivative at the
critical point
(which is an inflexion point of the critical isothermal) thus
boils down to the following:
0 = d/dV
( -
a / V2 +
2 a b / V3 )
Therefore: V = 3b
With this, we obtain p from the previous vanishing expression and
and T from the equation of state.
The critical point is thus characterized by:
Vc = 3b ,
pc = a / 27 b2 ,
Tc = 8a / 27 R b
Using those critical values as units of volume, pressure and temperature, the above
Van der Waals equation
takes on the following reduced form :
( p + 3/V 2 )
( V - 1/3 )
= 8T/3
The principle of corresponding states says that different substances at the
same reduced pressure and temperature will have the same reduced volume.
This fails for real fluids (especially at high pressure) but it's true of
any 3-parameter model similar to the Van der Waals model (see next).
The Dieterici equation of state is another 3-parameter model which predicts
a liquid-vapor transition and an associated critical state:
p (V-b) = RT exp(-a / RTV)
The innovative introduction of an exponential term was justified theoretically
by Conrad Dieterici (1858-1929).
The above predicts a
critical compressibility factor (Z = pV/RT)
of 0.27067, which is more realistic than the
Van der Waals value of 0.375.
In spite of this and other advantages,
the Dieterici equation remains far less popular than the Van der Waals equation.
Carnahan-Starling equation (1969) and beyond :
In 1969, Norman F. Carnahan (b.1942)
and K.E. Starling proposed the following 2-parameter equation of state,
expressed in terms of y = b/4V :
p V = RT
( 1 + y + y2 - y3 ) / (1 - y) 3
The CS equation is based on a model of rigid nonattracting spheres and directly
improves upon the aforementioned Clausius equation of state.
To account for the interaction between molecules,
the following 3-parameter equations have been proposed as refinements of the
Van der Waals and Dieterici
approaches, respectively:
( p + a / V2 ) V = RT
( 1 + y + y2 - y3 ) / (1 - y) 3
p V = RT exp(-a / RTV)
( 1 + y + y2 - y3 ) / (1 - y) 3
All equations of state featuring three parameters (a,b,R)
are identical for all substances, if stated in units of the critical temperature, pressure and volume.
g2hn (Yahoo!
2007-07-07)
How do I obtain the (molar) Van der Waals constants (a, b, R)
for ethane,
knowing the critical density is 0.21 g/cc
at tc = 32.1°C
and pc = 48.8 atm?
The mass of a mole of ethane
(C2H6CAS 74-84-0)
being 30.07 g the critical molar volume is
Vc = 30.07 / 0.21 = 143.19 cc.
Therefore, b = Vc / 3
= 47.73 mL = 0.00004773 m3.
Since pc = 48.8 atm = 4944660 Pa, we have
a = 27 b2 pc = 0.30415 J.m3.
Tc = 273.15 + 32.1 = 305.25 K gives
R = 8a / (27 b Tc ) = 6.18534 J/K.
In this context, R is just another adjustable parameter but it's
useful to check that its value is not too far from the
ideal gas constant :
8.3145 J/K.
(2006-11-11) Virial equation of state [molar]
pV = RT [ 1
+ B(T) / V
+ C(T) / V2
+ D(T) / V3
+ ... ]
This general expansion was proposed in 1885 by
Max Thiesen (1849-1936).
In 1901, H. K. Onnes (1853-1926;
Nobel 1913)
coined the name second virial coefficient; for B(T).
C(T) is the third virial coefficient, etc.
We're following the usual academic practice to equate the first virial coefficient
to unity.
For a "best fit", this would assume that the constant R may be adjusted at will...
In practice (especially when comparing several actual gases) it can be better to use the
standard value
of the gas constant for R and allow values of the first virial coefficient
which may differ (slightly) from unity.
Under the proper mathematical assumptions, the ith virial coefficient is ascribed
to those mutual interactions within a set of i molecules which
do not reduce to interactions within a smaller set
(in a way loosely reminiscent of
inclusion-exclusion enumeration).
An equation of state where all virial coefficients would vanish beyond the
second one would not be good enough to predict a liquid-vapor
phase transition.
A fluid so described could not have a critical state
(where the first and second isothermal derivatives of pressure over volume
vanish together).
Proving this is an interesting little exercise, which is left to the reader...
Boyle Temperature :
Real gases obey the Boyle-Mariotte law only approximately.
At low pressures (i.e., large molar volumes) the approximation is tightest for
a temperature which makes the second virial coefficient B(T) vanish.
Such a temperature is a characteristic of a given gas
called its Boyle temperature.
The Boyle temperature for a Van der Waals gas is thus:
TB = a / Rb
which is 27/8 = 3.375 times the
aforementioned critical temperature.
Experimentally however,
we're told
that the ratio of the Boyle temperature to the critical temperature
is consistently around 2.75.
The Dieterici equation of state gives a ratio of about 2.52 instead.
When starting from zero pressure at a constant temperature below the
Boyle temperature, the product pV first
decreases with pressure to reach a minimum, then it increases with pressure.
On the other hand, above the Boyle temperature, the product pV always
increases with pressure.
(2002-06-07)
What's viscosity?
A viscous deformation is a lateral deformation which occurs at a rate
that's proportional to shear stress.
Such a deformation is most readily observed with thick paste, but it occurs
much faster in not-so-thick liquids (low viscosity).
Technically, tar pitch
is actually a thick liquid of extremely high viscosity.
On the other hand, contrary to urban legends,
glasses are amorphous solids at room temperature.
They do not behave like "supercooled liquids" (unless heated
above a certain transition point) but they may "creep" like crystalline solids do.
For crystalline solids and/or rocks,
what occurs [over geological periods of time]
is a so-called "plastic" deformation.
The rate of plastic flow is not directly proportional to stress but
rather to some increasing function of stress with a zero derivative at the origin
(so that there's no plastic deformation at all when the stress is low).
For example, in what's called "Power Law Creep", the strain rate is proportional to
some n-th power of stress (n>1) and to a temperature factor exp(-Q/RT),
which indicates how easily crystal dislocations may move at an absolute temperature T.
For typical rocks, the exponent n is around 3 (it ranges from slightly below 2 up to
8 or so) and the "activation energy" Q is roughly 200 kJ/mol
(it may range from below 100 kJ/mol up to about 500 kJ/mol).
Viscous flow is a special case (n = 1 and Q = 0) of Power Law Creep...
Quantitatively, viscosity is the ratio of
shear stress to shear strain rate.
(These two are proportional in the case of what's called a Newtonian fluid.)
Units of viscosity are thus homogeneous to the product of a stress
(or pressure) by a time.
Therefore, the SI unit of viscosity is the pascal-second
(Pa×s), also called poiseuille (Pl),
which is 10 times as large as the corresponding [deprecated] CGS unit,
the poise (P) of 1 dyne-second per square centimeter.
The centipoise (cP) is a surviving synonym of the
millipascal-second (mPa×s),
which happens to be the viscosity of water at a temperature of about
20.2°C.
At 20°C, the viscosity of water is about
1.005 mPa×s and decreases with temperature:
At t°C, the viscosity in millipascal-second is within 0.2% of
1.005 exp(-0.024475(t-20) (1 - 0.8787
(t-20) / (t+98.5) )).
At 20°C, the viscosity of air is about
0.01808 mPa×s
and increases with temperature;
it's about 0.01709 mPa×s at 0°C.
The dynamic viscosity of gases
is roughly proportional to the
square root of the absolute temperature (T).
It changes very little with pressure.
Human blood
at 37°C is about 0.45 mPa×s
The above is called absolute or dynamic viscosity, as opposed to
the relative or kinematic viscosity, which is defined as the
quotient of the dynamic viscosity by the mass density,
expressed in units that are homogeneous to the ratio of an area and a time
(m2/s).
(Dynamic viscosity)(mPa×s
or cP)
=
(Kinematic viscosity)(cSt) * (Density)(kg / L)
The [deprecated] CGS unit of kinematic viscosity is the stokes (St),
which is 10000 times smaller than the SI unit
(m2/s).
The centistokes (cSt) is more commonly used in practice.
A kinematic viscosity is routinely obtained directly
with a [Saybolt or Engler] viscometer,
by timing the passage of a calibrated volume of liquid
under its own weight through a small aperture.
The reciprocal of [either flavor of] viscosity is called fluidity.
TransMineral USA. 2003-04-29)
Permeability :
Mortar made from our natural hydraulic lime
(NHL 5, with twice its volume of sand)
has vapor-exchange properties
Michel Couvreux (TransMineral USA. 2003-04-29)
Permeability :
Mortar made from our natural hydraulic lime
(NHL 5, with twice its volume of sand)
has vapor-exchange properties
listed
as 0.55 gram of air per hour,
per square meter, per mmHg [for one-coat renders].
What's its permeability?
Short Answer: 10 perm-inches (US) or
14.5 ng / s.m.Pa. Read on...
Note:
Besides the obvious problem with units, this question raises the issue
of converting a rating about the diffusion of air into the kind of water-vapor rating
which is clearly expected in the construction business.
We must also dispose of the fact that the stated rating is about a thickness of the
material which is not explicitely specified:
In the US, the thickness of "one-coat renders" is traditionally quoted as 3/8"
(there are normally 3 coats, including a thin outer coat, totalling 7/8" in thickness:
3/8+3/8+1/8).
However, the link quoted in the question makes explicit reference to a "French standard"
(which we've been unable to locate).
This seems to imply that "one-coat renders"
is shorthand for a metric reference thickness of 1 cm
(1 cm is only 5% more than 3/8").
We'll be assuming this much in the following article.
If you know better, please
let us know...
There are two kinds of permeability.
Geologists consider the flow of water or
oil
through rocks and deal with
hydraulic permeability (which we'll examine last) based on "Darcy's Law".
On the other hand, the diffusion permeability used in the construction industry
refers to "Fick's Law", the statement that the flux
of a diffusing substance is proportional to its concentration gradient.
For gases and vapors, partial pressures (more precisely, fugacities)
are used instead of "concentrations"... Air
and vapor ratings are different.
A layer of given thickness is rated according to permeance
(= permeability divided by thickness):
Permeance, for "Water Vapor Transmission" (WVT):
The permeance of a
vapor barrier
[more precisely, a "vapor diffusion retarder" or VDR]
is a measure of how fast it lets water vapor through,
when its two sides have different partial pressures of water vapor.
Permeance may be stated either in perms [US perms]
or in metric perms (which are about 52% larger than US perms).
These units are defined as follows:
1 US perm =
1 grain (gn) of water vapor per hour, per ft2, per inHg.
1 metric perm =
1 gram (g) of water vapor per day, per m2, per mmHg.
A perm is exactly 32399455 / 49161192, or about 0.659 of a metric perm.
Note that this "metric perm" unit
is not metric at all, since it's based on nonmetric units like the day
for time, and the millimeter of mercury (mmHg) for pressure.
The strict metric equivalents of both of the above units are:
57.213494670945101394- ng / s.m2.Pa
= 1 US perm.
86.812682389543557170+ ng / s.m2.Pa
= 1 metric perm.
The above SI unit (ng / s.m2.Pa) is homogeneous to a time divided by a length
and actually boils down to a "picosecond per meter"
(ps / m) but it's never expressed this way.
Unfortunately, this very metric unit has [wrongly] been dubbed
a "metric perm" by some sources...
The value of a perm in SI units does not depend on temperature !
Please ignore all those well-meaning Internet sources
[ 1 |
2 |
3 |
4 ]
which state otherwise by listing the above equivalences as valid at 0°C,
while giving slightly higher values (+0.418 %) for 23°C.
This nonsense comes from a poor understanding of units of pressure:
The millimeter of mercury (mmHg) and the inch of mercury (inHg)
are units based on the conventional density of mercury,
which is exactly 13595.1 g/L...
The fact that this is very close to the actual density of mercury at 0°C is
not directly relevant to unit conversions...
Even less relevant is the density at 23°C (about 13538.5 g/L)
which happens to be 0.418 % less [here we go].
This only means that, if we were measuring pressure with an actual column
of mercury at 23°C, we would have to divide the readings in millimeters by
about 1.00418 to obtain actual pressures expressed in so-called mmHg. Got it?
(If you didn't, you're in good company,
since even standardization organizations have been known to
introduce such silly "units" of pressures, whose values vary with temperature,
just like the actual densities of mercury and water do.)
Permeance is occasionally given in g/m2/day (DIN 53122) where a
"standard" vapor-pressure difference is understood,
which corresponds to a difference in relative humidity
of 85% at 23°C between both sides (for example, 0%||85% or 15%||100%).
As the
saturation
vapor-pressure over liquid water
(100% r.h.) at 23°C is about 21.080 mmHg,
the standard vapor-pressure difference is 17.918 mmHg,
making the above unit equivalent to about (1/17.918) of a metric perm,
or 0.084683 of a US perm
(the conversion factor is about 11.8).
"Water-Vapor" Permeability:
The above permeance is equal to the [diffusion] permeability of the material
divided by its thickness.
Permeability is a characteristic of the material itself,
which may be expressed in "perm-inches":
If a material has a permeability of one perm-inch,
a one-inch layer of it has a permeance of one perm,
a two-inch layer has a permeance of ½ perm, etc.
(For the record, units of permeability are homogeneous to a time,
and a perm-inch is about 1.45322 picoseconds.)
WVT Properties of Some Construction Materials
(perm = "US" perm)
(*) DIN 53122
rating in g/m2/day is for 18 mmHg
(85% difference in relative humidity at 23°C)
Note: Data was compiled from various sources and
may have been adjusted for self-consistency.
The mass diffusivity of water vapor in air (D)
is known to be 2.42 10-5 m2/s...
This is the coefficient to use in Fick's equationJx = -D (dC/dx),
which relates the mass flux (J)
to the gradient of mass concentration (C).
The partial pressure (p) of a gas is obtained from C by introducing the molar mass (M)
into the ideal gas law: p = RTC/M.
Thus we have: J = -(DM/RT) (dp/dx). In other words,
the diffusion permeability of ideal water vapor is DM/RT,
where R is the
molar gas constant:
8.314 472(15) J/K/mol.
In the case of water vapor (M = 0.018 kg)
diffusing in air at T = 300 K,
this gives a permeability of about 175 ng/s.m.Pa,
or 120 perm-inches, as shown in the first line of the above table.
Concerning NHL mortar, the unit given in Michel's question seems equivalent to
24 metric perms (since the time basis is an hour instead of a day),
so that 0.55 of such a unit would be exactly 13.2 metric perms
(about 20 US perms) if it was a true WVT rating.
Unfortunately it's explicitely specified as an air rating instead,
so we must somehow convert that into a water-vapor rating by considering
the different diffusion properties of air and water vapor...
If we assume that most pores in the mortar are much larger than the dimensions of the
molecules of the gases involved, we have microscopic
spaces in which the molecules bounce in thermal equilibrium,
in much the same way they would in an open space.
Under this simplification, each molecule of an ideal gas
of molar mass M would essentially diffuse
at a rate proportional to its average speed, which is itself proportional to
Ö(T/M) at temperature T.
Water vapor (H2O) has a molar mass of 18.0153 g.
Dry air is essentially 79% N2 (28.0135 g)
and 21% O2 (31.9988 g).
Ignoring interesting details, like the nitrogen enrichment of air diffusing
through mortar, we'll consider that
air is roughly a simple gas of molar mass 28.96456 g.
Such a gas would diffuse about 1.268 times slower than water vapor
(this is the square root of the molar mass ratio).
Accordingly, the permeance is roughly 1.268 times larger for water vapor
than for air, so the WVT permeance of a 1 cm layer
of NHL mortar is about 25.40 US perms (16.74 metric perms);
the permeability of 1:2 NHL5 mortar is thus
very nearly equal to 10 perm-inches.
Hydraulic Permeability
Darcy's
Law (of which there's a more general
tensor form)
states that a porous material lets a fluid through at a rate
inversely proportional to its viscosity
and directly proportional to both the cross-sectional area and the gradient
of the applied pressure (also called hydraulic gradient).
The material's [intrinsic] permeability is simply the coefficient of proportionality
in this law:
(Mark Barnes, UK.
2000-12-05)
What are the resonant frequencies of a given volume of enclosed air?
The amplitude U of a wave propagating at celerity V obeys the
wave equation :
1
¶ 2 U
=
¶ 2 U
+
¶ 2 U
+
¶ 2 U
V 2
¶ t 2
¶ x 2
¶ y 2
¶ z 2
=
DU
[D is the Laplacian operator]
As used above, the term "celerity" is best reserved for the "phase speed" which appears
in this equation. Hereafter, V is the
speed of sound in air.
The elementary solutions of this wave equation
whose amplitudes are zero on the walls of a rectangular box are obtained as the products
of single-dimensional solutions for a segment
(i.e., sine waves whose half-wavelengths are a submultiple of the segment's length,
so they are zero at both ends).
Such an elementary solution of frequency n
has therefore the following form for a rectangular box of dimensions A, B and C,
when the origin of coordinates is one corner of the box
(in this, a, b and c are integers):
U(x,y,z,t) = sin(apx/A) sin(bpy/B) sin(cpz/C) exp(2ip
n t)
The Laplacian DU is thus
-[(a/A)2+(b/B)2+(c/C)2]p2U
whereas the second time derivative of U is simply
-[4n2]p2U
.
As the above wave equation tells us, the ratio of those two square brackets
is equal to the square of the speed of sound V.
This gives us the following formula for
a resonant frequency (n) of air in a rectangular box
(where a, b and c are any positive integers):
n = ½V Ö
a2/A2 +
b2/B2 +
c2/C2
For a cylindrical box, say, you would solve a 2-dimensional equation for the disk
(using Bessel functions) and combine things as above into another formula
for n.
The lowest resonant frequency is, of course, obtained for
a = b = c = 1.
The next frequencies are not multiples of this one...
The general solution of the wave equation for the cavity is a superposition
of the above vibrations (à la Fourier).
We assumed perfectly rigid walls.
The relative correction for vibrating walls will typically be as small as the ratio
of the vibration amplitude (probably less than a millimeter)
to the wavelength in air (about a meter at 343 Hz, for air at 20°C)
The notion that resonant frequencies are directly related to volume is a myth,
but we may investigate what happens to the lowest resonant frequency
(a = b = c = 1)
for a constant volume L3 = ABC
(A = xL, B = yL, C = L/xy):
n =
no Ö
( 1/x2+1/y2+x2y2 ) / 3
This happens to have a minimum at
n = no
in the case of the cube (x = y = 1).
For a cavity that's not too different from a cube
(which may not help much with guitar design)
n is thus pretty close to
½Ö3 V/L,
where L is the cubic root of the volume, and V is the
speed of sound...
How the pitch of wind instruments vary with temperature :
To conclude, we may examine temperature dependency:
As the thermal dilatation of the instrument itself is very minute,
the frequencies are essentially proportional to the
speed of sound V,
which is itself proportional to the square root of the absolute
temperature T (for an ideal gas).
At room temperature (T = 295 K),
this means that a variation of one kelvin (1°C)
will induce a frequency change of about 0.17%.
This translates musically into (take your pick):
0.00244 octave, 0.0293 semitones, 0.735 savart, 1.465 centitone,
2.93 cents or 2.44 millioctaves.
That's not much but it can be noticeable:
It takes a difference of about 10°C for wind instruments
to be off by 30 cents (30% of a semitone).
Wind instrumrnts in a warm interior thus play about one semitone sharper
than they might in freezing weather outdoors. (Strictly speaking,
this applies only to organ pipes which use ambient air,
not to smaller wind instrument whose inside air is constantly replenished
by warm human breath.)
In an orchestra, string instruments are affected the other way
(the warmer the flatter) mostly because string tension decreases notably
even with the minute increase in length due to
temperature dilatation. They can adjust (violins) and/or retune (guitars)
so everybody can stay in tune.
Pianos are a problem, keep them indoors...
(J. L. of Canada. 2000-11-23)
The Whole Atmosphere
The atmosphere is made of molecules, piled up on top of each other.
It starts at earth's surface and becomes indistinguishable from the vacuum
of space a few hundred kilometers up.
Now, consider a 1 cm by 1 cm square sitting at sea level and all the air that's
sitting on top of that square, from the surface all the way up to outer space.
What is the approximate mass of the air in that one square-centimeter column?
The short answer is that pressure comes from the weight of the column of air
above the surface. The standard atmospheric pressure of 101325 Pa exerts a force of
10.1325 N on a surface of a square centimeter.
The problem is to estimate the
mass from the weight. As we shall see, Earth's gravity does not vary
much for most of the atmosphere, so we may use the standard value of gravity
(9.80665 m/s2 ) and come up with a mass of
approximately 10.1325 / 9.80665, or roughly 1.033 kg
over each square centimeter at sea level.
Indeed, Earth's gravity is approximately constant throughout the atmosphere:
At an altitude equal to 0.5% of the Earth radius (about 32km),
it is only 1% less than at sea level, and most of the atmosphere
is below that point (more than 99% of it is said to be below an altitude of about 70 km,
where gravity is only down 2%).
Consider a column of air at rest (it's essentially part of a slim cone whose apex is at
the center of the Earth).
The weight of all the molecules of air ultimately results in a single force pS exerted
by the pressure p at sea-level on the surface area S at sea-level.
Now the weight (that's force, not mass) of each molecule of mass m is
slightly less than the weight at sea level mg (g being gravity at sea-level)
so that the mass is slightly more than pS/g,
but not much more, as previously observed.
Taking the standard values g=9.80665 m/s2 and p=101325 Pa,
a surface
S=1 cm2= 0.0001 m2 would have approximately 1.03323 kg of air above it.
As we've seen, this is actually a slight underestimate of the true value,
so we may state that a cm2 at sea-level has
slightly more than a kilogram of air above it.
The interesting thing is to estimate the total mass of the atmosphere.
The surface of the Earth's is very close to
5.1 1014 m2
(that's the surface of the reference ellipsoid and that's also very close to the
surface area of a sphere whose radius is the "conventional" radius of the Earth,
namely 6371 km).
Therefore, the above gives a total mass of about
5.27 1018 kg.
Now there's a problem: the mass of the atmosphere is listed as
5.136 1018 kg in
my copy of the 1995 CRC Handbook of Chemistry and Physics (page 14-7).
We should not have been troubled at all about the small (2.6%) discrepancy
from our crude estimate, except that it turns out to be in the wrong direction:
If the variation of gravity with altitude was the only factor to correct,
the true number should be higher, not lower.
You may remark that more air is packed at the poles
(the temperature's lower, the density's higher) with a larger surface gravity
(9.832186 m/s2 at the poles).
This makes a correction in the right direction, but it cannot exceed 0.26%
(that's how much the gravity at the pole exceeds the standard value of g)
whereas our discrepancy is 10 times as large.
What else could be wrong?
Well, our value of the "standard" atmospheric pressure was precisely designed
to make this particular computation work out right and it is not at fault...
The reason for the discrepancy is that we overlooked land masses in the computation!
Whatever volume is occupied by land is not available for air and does
decrease the total mass of the atmosphere.
To get a 2.6% difference in the mass of the atmosphere,
we would need the average land (30% of the surface of the globe)
to decrease by about 9% the mass of air above it, as compared to sea level.
That looks like a pretty reasonable figure, and so does the CRC value of
5.136 1018 kg
for the mass of the entire Earth's atmosphere.
The molar mass of air is about 28.966 g/mol, so there are about
1.773 1020 moles in the
atmosphere, that's a grand total of about
1.068 1044 individual
molecules. The square root of that is
1.033 1022, which is
0.01716 moles, or the number of molecules in 0.497 g of air.
This occupies a volume of about 400cc at 11°C under 1 atm.
In the folklore of physics, this volume was once known as "Caesar's last breath".
This volume is about equal to a human breath,
so each time we inhale, we take in about one of the molecules that Julius Caesar
last exhaled more than 2000 years ago (the 2000-year delay implies that
air has been thoroughly mixed since then).
This is so because the whole atmosphere is to a human breath (400cc) what a human breath
is to a single molecule.
We could spoil everybody's fun by pointing out
that quantum mechanics
tells us that identical molecules cannot be
distinguished, even in principle, so that the whole concern is
really fallacious (you just can't paint a molecule red)...
(2016-07-14) Composition of dry air at sea-level
Surveyors assume the concentration of carbon dioxide to be 450 ppm.
Calculating the average "molar mass of air" (28.966 g/mol)
from its main constituents at sea-level is fairly delicate.
The value used on Numericana is the sum of the last column
in the following table, properly rounded:
Simplified composition of dry air at sea-level :
Gas
mol/molair
g/mol
g/molair
N2
0.78076
28.0134
21.8717
O2
0.20944
31.9988
6.70183
Ar
0.009322
39.9481
0.372396
CO2
0.000450
44.0095
0.0198043
Ne
0.00001818
21.1797
0.00036687
He
0.00000524
4.026
0.0000209736
CH4
0.00000222
16.04
0.0000356088
Kr
0.00000114
83.798
0.0000955297
H2
0.00000055
2.01588
0.00000110873
N2O
0.00000031
44.013
0.000013644
CO
0.00000020
28.01
0.000005602
Xe
0.000000087
131.293
0.0000114225
O3
0.000000040
47.9982
0.00000191993
NO2
0.000000020
46.0055
0.00000092011
SO2
0.000000010
64.0638
0.000000640638
NH3
0.000000003
17.0305
0.0000000510916
Air
1.000000000
28.966
28.9663256
The above is a simplified version of the actual composition of the atmosphere
(the complex equlibrium between nitrogen oxides and ozone is summarized by
two species of fixed concentrations).
It has been adjusted to make the rounded fractions
have an exact sum of 1,
while matching the standard molar concentration of 450 ppm
for CO2 quoted by geodetic surveyors.
The above yields a value of 28.9663 g/mol for air.
(another "standard" using slightly different values, including 397 ppm
for carbon dioxide, yields 28.9657 g/mol for air).
(2016-08-03) Humidity. Moisture content of
clear atmospheric air.
Relative humidity, absolute humidity and specific humidity.
Moist air can be viewed as a mixture of water vapor and dry air
(consisting itself of fixed molar fractions of all other atmospheric gases,
including carbon dioxide).
The molar ratio of those two components is very nearly
equal to the ratio of their respective partial pressures.
Dalton's law (1801)
states that the sum of the partial pressures is equal to the total pressure
measured by an ordinary barometer.
Relative Humidity (RH, j) :
At some given ambient temperature T, the partial pressure of
water vapor pv
cannot exceed the saturation pressure p*(T)
at which vapor would condensate into liquid droplets (as dew, fog or clouds).
The composition of clear moist air is thus most commonly specified in terms
of its relative humidity (RH) defined as the following
ratio, which can vary between 0 and 1
and is usually expressed as a percentage (0 to 100%).
(2008-06-04) The first hot-air balloon flew 225 years ago.
First public demonstration of a Montgolfière : June 4, 1783.
Joseph Montgolfier (1740-1810) and Etienne Montgolfier (1745-1799)
were two of the 16 children of a wealthy French paper manufacturer.
On June 4, 1783, they made the first public demonstration of an hot-air balloon
which they privately called Seraphina.
It was 11 m across and its sections were held together by more than
2000 buttons...
The balloon was not tethered and had no payload but it was intended as
a prototype of an aérostat
large enough to take people into the air.
Seraphina rose more than 1000 m in the sky of
Annonay,
before landing in a vineyard about 2 km away from the take-off point.
That event was
commemorated worlwide by Google on June 4, 2008, with the following logo:
The first manned untethered flight, however,
occurred several months later:
On November 21, 1783,
a silk balloon made by Etienne Montgolfier
took off at 13:54 pm, carrying two people aboard a wicker nacelle:
the physicist
Jean-François Pilâtre de Rozier (1754-1785)
and the marquis
François-Laurent d'Arlandes (1742-1809).
They flew
from the Jardins de la Muette to
la Butte-aux-Cailles in Paris
(a distance of about 9 km).
The flight lasted 28 minutes and an altitude
of 960 m was reached. The balloon had
a height of roughly 23 m, a diameter of 15 m
and a volume of 2200 m3.
A balloon of that size would contain about 2800 kg of air.
So, by Charles' law and Archimedes' principle,
280 kg would be lifted by raising the temperature of that air
10% (roughly, 30°C or
54°F above atmospheric temperature).
At first, the Mongolfier brothers thought that burning wool and straw
released an unknown
light gas (dubbed "gaz Montgolfier")
but the physicist Horace de Saussure
(1740-1799) pointed out that the effect was purely thermal.
The Montgolfier brothers had
experimented with hydrogen
(the newly discovered "flammable air")
but could not design a membrane impermeable to hydrogen.
The erroneous early reports that their first successful balloons were filled with a light gas had
one happy consequence: The physicist Jacques Charles (1746-1823) rushed
to "reproduce" their success and came up with the first successfull hydrogen
balloon shortly thereafter (see below).
The official report for the French Académie des Sciences
was written by Benjamin Franklin.
Another report
was written by d'Arlandes himself.
Less than two weeks later
(on December 1st, 1783) Jacques Charles, accompanied by
Marie-Noël Robert, would fly from the Jardin des Tuileries with a
more practical balloon
(dubbed La Charlière)
which had been crafted by the brothers Jean and Marie-Noël Robert.
It was a much smaller balloon inflated with hydrogen and
its envelope was made of silk impregnated with elastic gum.
Dirigible balloons were conceived shortly thereafter (1784) by a
brilliant mathematician and future revolutionary
general, Jean-Baptiste
Meusnier (1754-1793)
who is still remembered for his studies of minimal surfaces
and of lines drawn on a curved surface
(Meusnier's Theorem, 1776).
The gas-balloon technology pioneered by Charles quickly supplanted hot-air balloons
(which were only revived in the late 1950s).
Yet, on one glorious day of 1783, it's the
montgolfière which captured imaginations by allowing
Rozier and d'Arlandes to become the
first humans to fly !
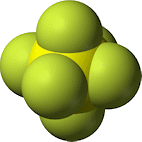
(2011-07-31) Sulfur Hexafluoride (SF6 )
A very heavy gas and a good electrical insulator.
Under atmospheric pressure, sulfur hexafluoride
is a colorless gas above 209.3 K (-64°C).
Its molar mass of
146.055 g/mol.
makes it 5 times heavier than air.
Being stable and non-toxic,
sulfur hexafluoride
is safe to use in
popular demonstrations,
unlike the following heavier gases:
In the atmosphere, sulfur hexafluoride has a lifetime (life expectancy)
of 3200 years (that's a half-life of 3200 ln 2 =
4600 years).
It's the most potent greenhouse gas
ever evaluated by the
IPCC:
On time scales of 20, 100 and 500 years, sulfur hexafluoride is respectively
16300, 22800 and 32600 times more potent than carbon dioxide.
In spite of its low current abundancy, the gas thus contributes to an estimated
0.2% of the observed global warming and it's banned in Europe for all industrial uses,
except in high-voltage
gas-insulated switchgear (GIS)
where it's irreplaceable.